Overview
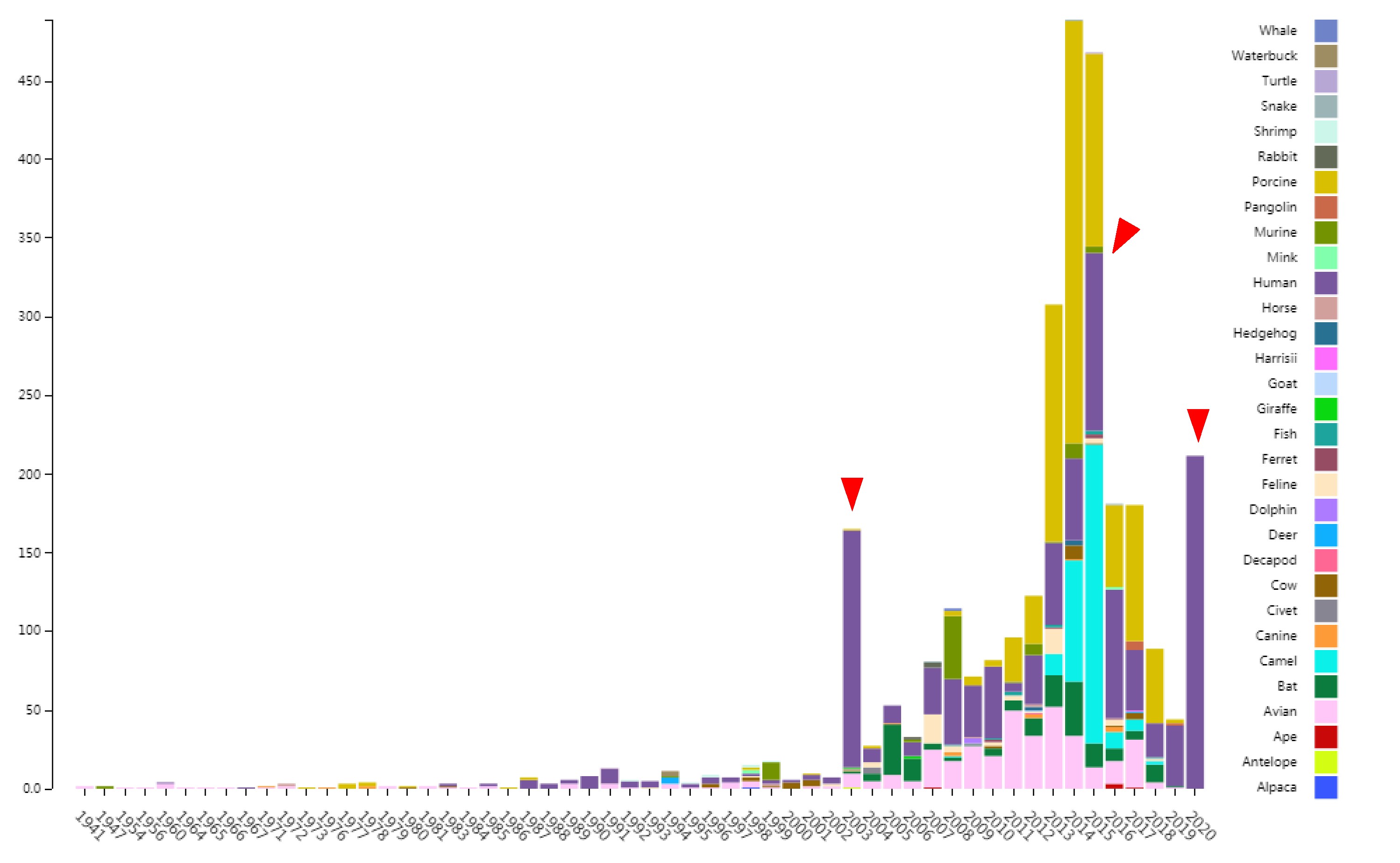
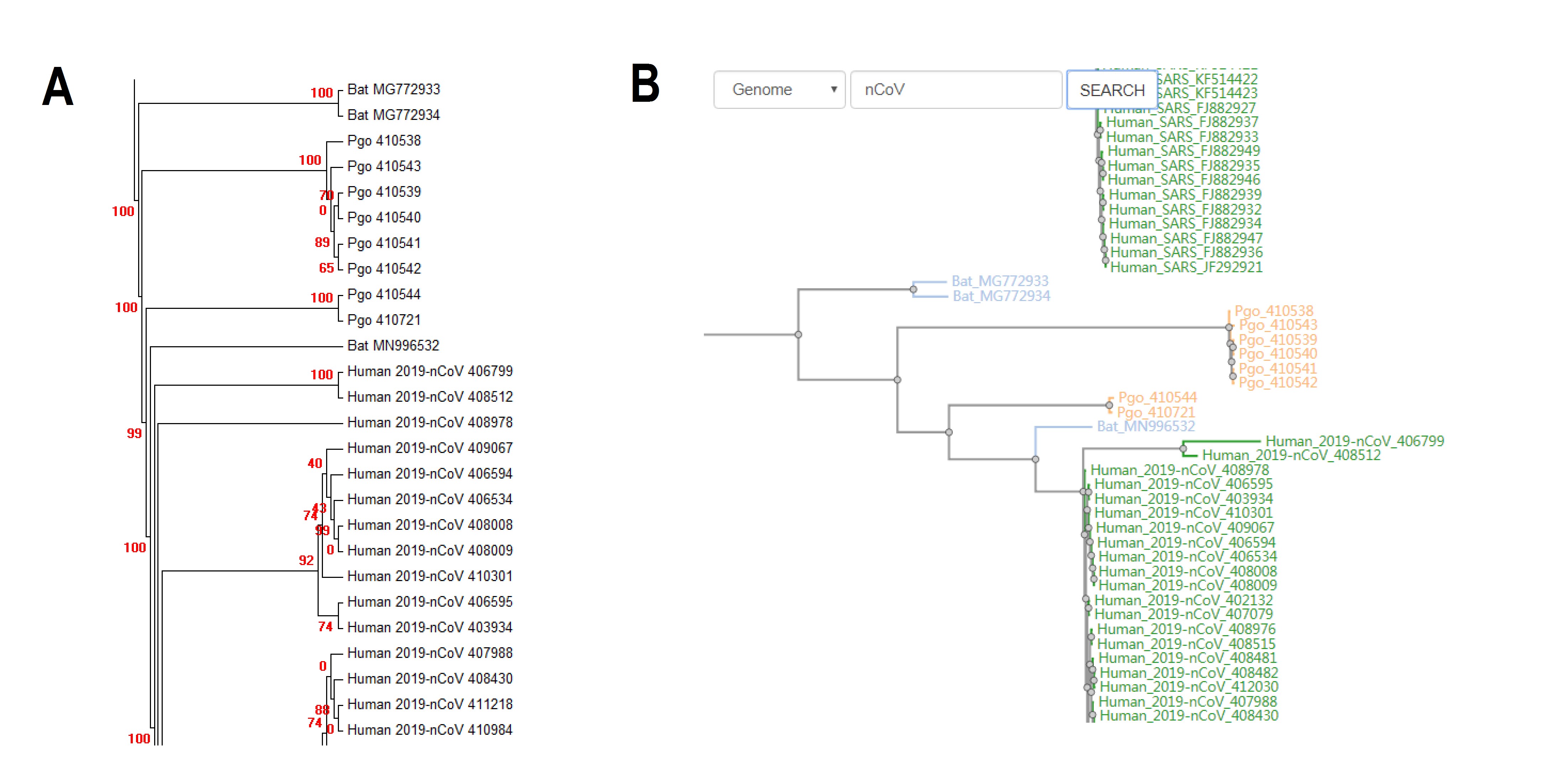
CoVdb extensively collects published coronavirus data and have taken in genomes of 2965 strains, which were collected from 31 organisms and in the years from 1941 to present, 2020 (Figure 1). 972 (33%) are human coronavirus and 176 (6%) are bat coronavirus, which are referred as the possible source of human coronavirus. Porcine coronavirus also take a big percentage (822, 28%) and coronavirus used to make damages in the pig industry. In time series of the number of sequenced human coronavirus, there are three peaks, which represent the outbreaks of SARS-CoV in 2003, MERS-CoV in 2014-2015 and 2019-nCoV in 2019-2020, separately. Using all documented coronavirus genomes in CoVdb, we generated a phylogenetic tree of coronavirus (Figure 2A), from which we observed that the nearest non-human strain of 2019-nCoV is Bat_MN996532 (Bat-CoV-RaTG13), a strain isolated from Rhinolophus affinis, a species of bat in the Rhinolophidae family. Pgo_410721 is also in the vicinity of 2019-nCoV and its host, pangolin, was once considered as a potential intermediate host of 2019-nCoV. We developed search tools to allow users to swiftly find a strain in a big phylogenetic tree (Figure 2B).
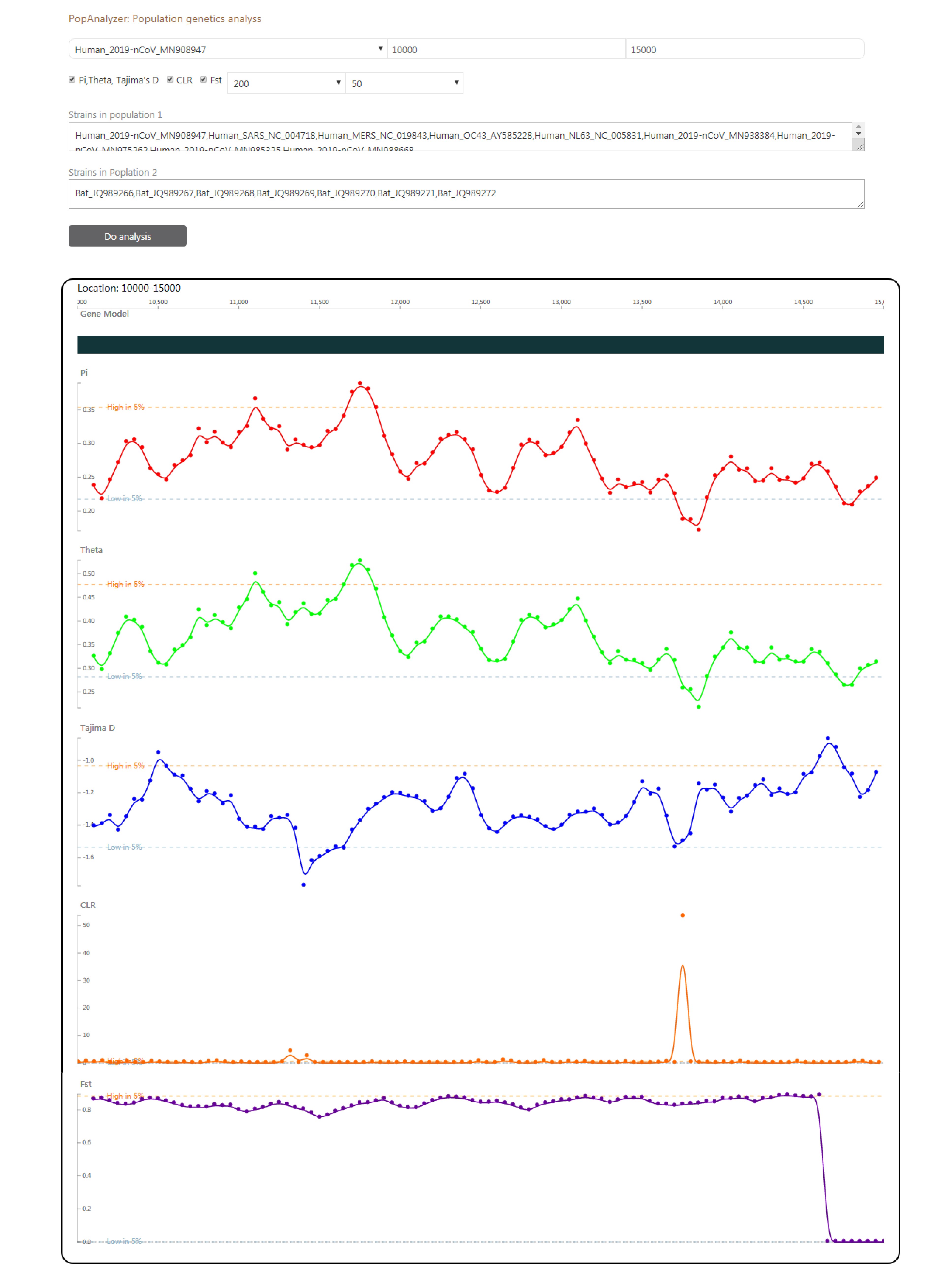
In average, there are 5-14 possible open reading frames (ORF) or genes in one coronavirus strain. We grouped homologous coronavirus genes (requiring identity > 0.5 and coverage > 0.8) into 628 clusters (for details, see Materials and Methods). This number indicates that the differentiation or diversity within coronavirus isolates is not low. For these, we still performed a subcellular localization analysis for the 628 clusters to predict their roles in infection, although the structure of coronavirus is not complex. Base on prediction only, 21% (133 items) are predicted to be located in the host nucleus or host cytoplasm, while 40% (250 items) are predicted to be membrane proteins (Figure S1). In gene ontology, using WEGO, we found coronavirus genes enrich in association to the membrane. For general information, CoVdb includes 13471 function annotations and 155942 GOs. We searched for the closest protein structure of each coronavirus genes in the Protein Data Bank (PDB), and acquired 299105 mappings with a E-value < 0.05 and a coverage > 50%. For all human coronavirus strains, we did sliding window analyses for Pi, Tajima’s D, composite likelihood ratio (CLR) and fixation index (Fst). For Pi, Tajima’s D and CLR, the target group is one specific human coronavirus, such 2019-nCoV, MERS or SARS, or coronavirus of some specific host, such as human coronavirus, bat coronavirus or porcine coronavirus. Fst is between human coronavirus and one non-human coronavirus. All these data can be viewed in CoVdb’s genome browser.
Main Functions
The genome browser (GBrowser) in CoVdb follows a style with analysis tracks (CLR, Pi, Tajima’s D, and Fst) listed following gene segments. CoVdb also are equipped with other general genome browser tools, such as zoom in/out or position movements. In addition to basic information, CoVdb show gene information mainly in function annotation, subcellular localization, topology and protein structure. The search engine in CoVdb is powerful and supports fuzzy search, BLAT and BLAST. CoVdb also allows to search by cell location. For personalized analyses, CoVdb is able to provide gene links if inputting a list of chromosome positions or gene accessions.
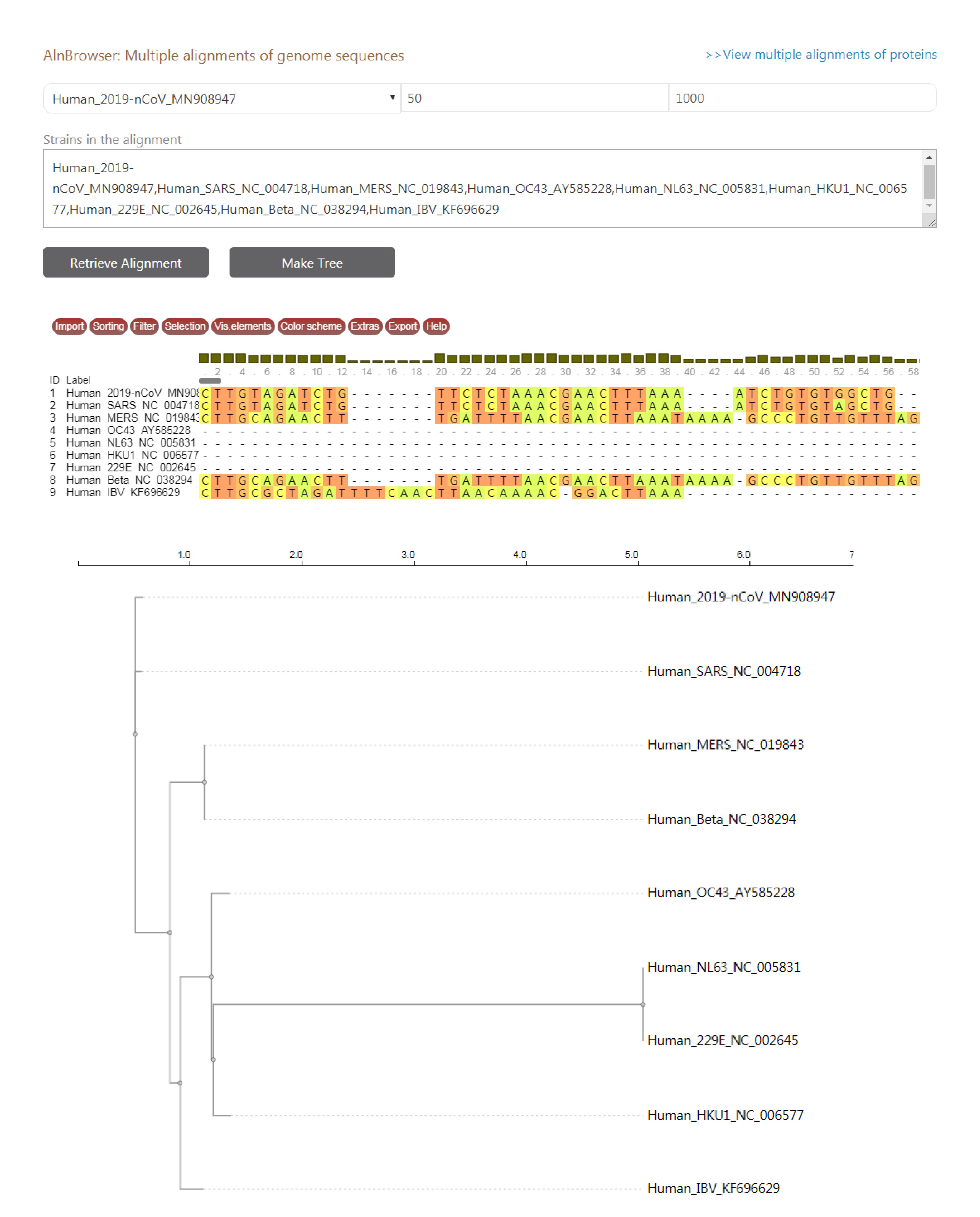
CoVdb has tools to facilitate some specific use in coronavirus research, such as tracing origination, vaccine or drug design. In the tool “Protein”, genes’ 3D structure related information are listed, in which users can view the overlapped amino acids of a coronavirus protein in its 3D structure counterpart and do online protein structure analyses. Users also can search a protein sequence and view the mapped region in the target’s 3D structure. The tool “AlnBrowser” allows users to retrieve multiple alignment of two or more strains at some position and build a phylogenetic tree using the alignment (Figure 3). With the tool “PopAnalyzer”, users can do personalized online sliding window analyses. Users can choose the window size, the step size, the target region and the population genetic tests (Figure 4). “Phylo Tree” is a tool to view and search in phylogentic trees made by genomic or proteomic sequences (Figure 2B). Users also can go to the GBrowser page by clicking the name of one strain.